|
Our recent effort in mitochondrial DNA (mtDNA) sequencing and quality control resulted in the development of a forensic mtDNA database, mtDNAmanager (http://mtmanager.yonsei.ac.kr). With this bioinformatics resource, we expect to provide a guideline for stringent quality control of mtDNA data to eliminate most systematic errors (e.g., a mix-up of site designations, base shifts or mistypings) using a new approach based on haplogroup estimation and data comparison with previously reported high quality mtDNA sequences. Another recent research interest in our lab is the development of genotyping methods which enables the detection and analysis of genetic characteristics of highly degraded DNA, such as in old skeletal remains. To increase the PCR efficiency in these conditions, efforts to extract DNA with high yield and to design PCR systems to yield small amplification products have been made.
Mitochondrial DNA |
| Analysis of the human mitochondrial DNA control region has become a powerful tool for forensic identity testing. Patterns of mutations which have accumulated over time along the transmission of mtDNA lineage give rise to the differences among individuals, and have become the basis for forensic discrimination. Other properties of mtDNA that make it valuable for human identification studies include its high copy number, maternal inheritance, and the absence of recombination. Thanks to these outstanding features, mtDNA has also been widely exploited in various disciplines, such as population, medical, and forensic genetics. However, the current population, medical and forensic genetic databases still have a large number of flawed sequence data in spite of several suggestions which have been made for avoiding errors in mtDNA sequencing and documentation. The recent interest and availability of applications for detecting errors in mtDNA data has resulted in the mtDNA haplogroup determination based on the well characterized mtDNA phylogeny as a useful a posteriori check on the quality of sequence data. In this regard, we collected 593 Korean mtDNA control region sequences of high quality and determined their corresponding haplogroups by confirming the coding region SNP information. This method allowed the complementation of coding region information with control region mutation motifs and the resultant findings suggest reliable control region mutation motifs for the assignment of East Asian mtDNA haplogroups. Moving forward, we identified the reliable control region mutation motifs for the assignment of more than 380 mtDNA haplogroups and subhaplogroups based on the well characterized global mtDNA phylogeny. Then, to support mtDNA data quality control based on haplogroup estimation and database comparison, we developed a web-based forensic mitochondrial bioinformatics resource, mtDNAmanager (http://mtmanager.yonsei.ac.kr). The mtDNAmanager consists of previously reported high quality mtDNA sequences (7,090 mtDNA control region sequences from various world-wide populations in April, 2008), and a set of bioinformatics tools, able to automatically characterize newly submitted data by estimating its haplogroup according to the haplogroup-specific control region mutation motif. However, as many of polymorphisms by which mtDNA haplogroups can be defined are specific to populations, we will keep expanding database for some time to contain more world-wide mtDNA sequences of high quality. |
 |
--------------------------------------------------------------------------------------------------------------------------
Y-chromosomal STR |
| STRs on the Y chromosome have been very useful, not only because of their application to forensic studies, but also because of their use in studies of human migration and evolution. Since the human Y-STR markers are haploidly inherited without recombination, the allele combination of a series of individual Y-STR loci has thus been considered a haplotype. The more highly polymorphic markers are added to the established Y-STR core set, the higher potential to distinguish different paternal lineages can be achieved. However, highly polymorphic STR loci may have high mutation rates because there is natural correlation between the degree of polymorphism and the mutation rate of a given locus. In paternity testing, spontaneous mutations in the germline of the putative father at any locus used in the analysis can lead to a false exclusion due to differences between father and son. Therefore, reliable estimates of mutation rates of Y-STRs are also necessary for the accurate interpretation of Y-STR data in paternity testing and forensic casework. In this regard, we analyzed 22 Y-specific STR loci (DYS19, DYS389I/II, DYS390, DYS391, DYS392, DYS393, DYS385, DYS388, DYS437, DYS438, DYS439, DYS446, DYS447, DYS448, DYS449, DYS456, DYS458, DYS464, DYS635, and GATA H4.1) in 708 unrelated Korean males, and evaluated the usefulness of these markers. Then, we analyzed 369 Korean father/son haplotype transfers in 355 families at the 22 Y-STRs to calculate mutation frequencies. During these analyses, we also found that DYS385 shows allele size differences depending on the primer set and that DYS448 deletion occurs associated with arrangements of the azoospermia factor C gene (AZFc). Recently, mini-STR PCR designing and commercializing effort are also being made for the Y-STR analysis in old skeletal remains. |
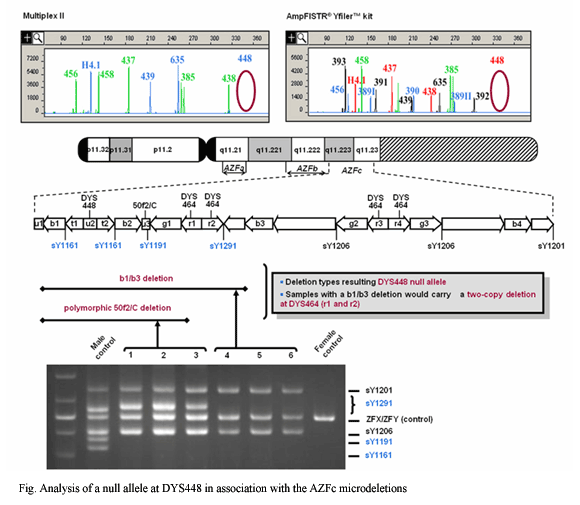 |
--------------------------------------------------------------------------------------------------------------------------
X-chromosomal STR |
| It became clear that the unique mode of inheritance of the X-linked markers and the intermediate state of the X-chromosome between diploid and haploid chromosomes bears the potential for haplotyping and, accordingly, for solving complex kinship cases. In deficiency cases, X-chromosome linked STRs became a powerful tool especially when the disputed child is female. X-STRs are so more efficient than autosomal STRs in these cases that some special deficiency cases can only be solved through X-linked markers. Therefore, we selected and analyzed nine highly informative X-STRs (HumARA, DXS9898, DXS6809, DXS101, DXS7424, GATA172D05, HPRTB, DXS8377 and DXS10011) in 150 male and 150 female Koreans to show the polymorphisms of these markers and to evaluate their efficiency in forensic practice. We found that the nine X-STRs are very informative for forensic use and that no linkage disequilibrium exists among these nine STRs. Recently, haplotype stability as well as linkage disequilibrium among several X-STRs (DXS8378, HPRTB, DXS7423, DXS7132, DXS10134, DXS10074, DXS10101 and DXS10135) and their differences between populations became new fields of special interest. Therefore, we are now considering the research for these X-STRs in relation to ChrX haplotyping in Koreans. |
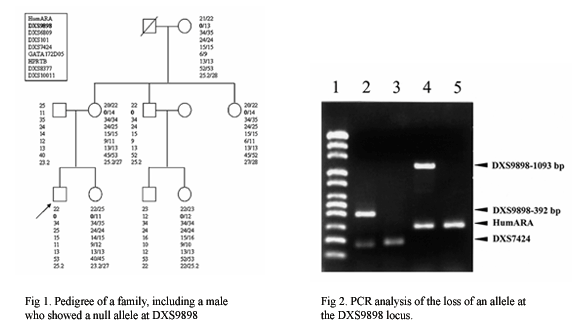 |
--------------------------------------------------------------------------------------------------------------------------
Degraded DNA |
| DNA samples from forensic cases are often degraded or tainted by environmental contaminants. When such low quality DNA is used for STR profiling, loss of signal is typically observed with larger-sized amplicons. One approach for recovering information from degraded samples is to reduce the PCR amplicon sizes by moving primers as close as possible to the STR repeat region. The observation that smaller-sized amplicons from autosomal STR markers produce a higher success rate with degraded DNA was confirmed by a number of recent studies. In this context, we expected that using reduced-size amplicons to type Y-STRs could also produce a high success rate with highly degraded forensic or anthropological samples. Therefore, we developed two Y-miniplexes (mini Y-STR multiplex PCR sets) by reducing the amplicon sizes of 21 Y-STR loci. The two Y-miniplexes included 17 Y-STR loci (DYS19, DYS385, DYS389I/II, DYS390, DYS391, DYS392, DYS393, DYS437, DYS438, DYS439, DYS448, DYS456, DYS458, DYS635, and GATA H4.1) from a commercial multiplex kit, AmpFlSTR® Yfiler™, and another four Y-STR loci (DYS388, DYS446, DYS447, and DYS449) to increase the haplotype diversity. We compared the effectiveness of the two multiplexes with the AmpFlSTR® Yfiler™ kit by using both enzymatically degraded DNA and 30 samples of 50-year-old skeletal remains. This comparison demonstrated that the new Y-miniplex sets can produce a better signal from degraded DNA than the AmpFlSTR® Yfiler™ kit. We think that using these miniSTRs will be helpful for identifying missing persons from mass disaster or civil wars, e.g. the Korean War (1950-1953) and are making effort to identify missing casualties and return them to their families on the basis of circumstantial evidence and matching results of mtDNA and autosomal- and Y-STR genetic profiles. Now, we are interested in the interpretation of DNA profiles obtained from highly degraded samples using expert systems. |
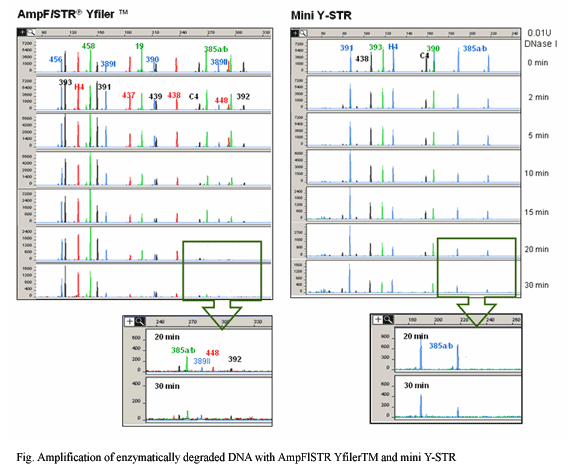 |
 |
|
